

In April, Ipsen’s Ojemda (tovorafenib) secured European Commission (EC) approval for the treatment of relapsed or refractory paediatric low-grade glioma. Notably, this approval is the first to initiate a 30-day countdown for the official endorsement of a Joint Clinical Assessment (JCA) report, a harmonised process designed to allow EU member countries to review data in parallel.
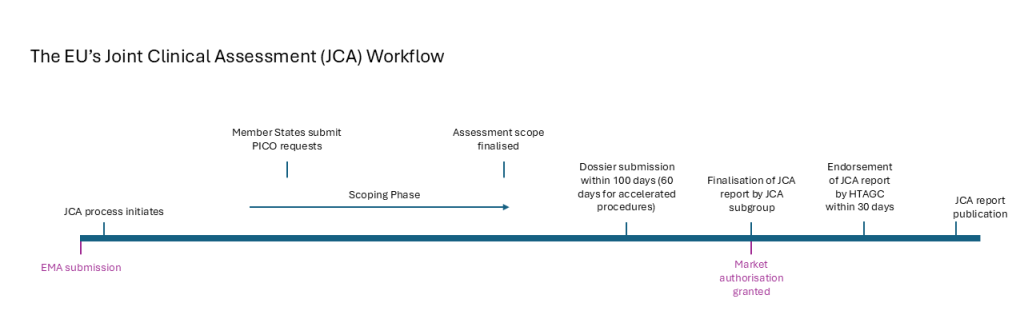
The JCA process was launched in January 2025 by the EC with the aim of streamlining clinical evaluation to accelerate access to innovative treatments. While currently applicable for oncology treatments and advanced therapy medical products (ATMPs), JCAs will be conducted for all other products in the coming years.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
Nonetheless, questions linger on the conservative approach outlined in the JCA guidelines to non-randomised evidence, the openness of communication channels, process-related complexities, and resource pressures, making it unclear how fit-for-purpose the JCA is in its current form.
What has changed under the JCA?
Under the new Health Technology Assessment (Regulation [EU] 2021/2282), or HTAR, the JCA subgroup, a dedicated working group within the Member State Coordination Group on Health Technology Assessment (HTACG), appoints an assessor and co-assessor from the EU member states to assess the relative clinical benefit of new technologies. It aims to replace multiple duplicative efforts by Member State HTA bodies by creating a single, centralised scientific report that can inform reimbursement and pricing decisions on a national level. However, the EC states that JCA will not provide “any value judgements or conclusions on reimbursement”, and member states would remain responsible for conducting cost-effectiveness evaluations based on regional needs.
Cancer Patients Europe’s ‘What is the Joint Clinical Assessment (JCA)?’
Industry experts have always had a positive perspective of the JCA approach and its aim to provide a “one-stop shop” for clinical assessment, says Dr Alexander Natz, Secretary General of the European Confederation of Pharmaceutical Entrepreneurs (EUCOPE). Similarly, Dr Sreeram Ramagopalan, Senior Lecturer at the Centre for Pharmaceutical Medicine Research, King’s College, London, UK, agrees that the objective to harmonise clinical assessment is valid, as varying HTA requirements have led to fragmented patient access across Europe.
However, the current approach is diverging somewhat from its original goal, explains Natz. HTAR was developed by the EC and Member States over several years and went through numerous iterations, says Patrick Hopkinson, founder and managing director, Wokingham, England-based PHTA Consulting. Its eventual form fell short of industry expectations, particularly regarding the “voluntary” rather than “mandatory” use of JCA reports by national HTA bodies, he adds.
Procedural concerns over JCA
The JCA process occurs in parallel to the EMA’s review of a drug’s MAA submission. First, the JCA scoping process kicks off with Member States defining their national clinical evaluation parameters, structured by population, intervention, comparator and outcomes or PICO. The assessor and co-assessor are responsible for providing a consolidated set of PICOs that define the parameters of the assessment.
A health technology developer must submit a dossier within 100 days of scope confirmation by the JCA subgroup. This is a tight timeline, which poses a substantial challenge for a developer, says Hopkinson. Anecdotally, Natz described dossiers as being up to 30,000 pages long, which places a substantial burden on resources, particularly for smaller companies.
These short timelines are particularly challenging due to the complexity of evidence required, notes Hopkinson. An increased risk of avoidable mistakes in the dossier preparation may jeopardize a product’s review outcome despite clinical benefit, says Natz.
Ipsen and Iovance Biotherapeutics, the second company expected to undergo a JCA for its product, did not provide comment for this story.
Challenges have emerged because Member States are requesting several different PICOs, which necessitate complex analysis, says Hopkinson. The choice of comparator products is one such example, explains Natz. While the standard of care may vary between countries, he says that there is a risk of overburdening developer resources and diluting the objective for a unified report. He calls for a balance to be struck between the needs of Member States and evidence requirements.
PICO requests can sometimes be politically driven, according to Natz. While comparator products should reflect the standard of care, they are sometimes selected with the goal of bringing down prices, he explains. While an open dialogue exists on a national level, the opportunity for discussion is currently limited within the JCA process, notes Matias Olsen, Senior Manager, Public Affairs & Policy, EUCOPE.
If different PICOs are requested, developers will not have sufficient time to generate the relevant data, which may be why some JCAs have been delayed, says Ramagopalan. Due to the tight timelines, companies are required to prepare a lot of the evidence “at risk” before scope confirmation, says Hopkinson, with some developers predicting comparative data requirements related to PICOs a year or two in advance, adds Ramagopalan.
Stringency of evidence requirements
Evidence generation for the JCA is based primarily on the German HTA approach, which has very stringent standards, including being critical of real-world evidence, single-arm studies, surrogate endpoints, and indirect treatment comparison, says Hopkinson.
The EC’s JCA guidance states that evidence generated outside of a randomised controlled trial (RCT) has “much greater potential to include material bias in the estimate of treatment effect”.
For some rare and ultra-rare diseases, which have a limited number of patients, an RCT is not feasible or ethical to conduct, placing more reliance on real-world data, says Paolo Morgese, Vice President of Public Affairs Europe for Alliance for Regenerative Medicine. The JCA process is due to be expanded to include orphan drugs in 2028, followed by all other new medical products in 2030.
Morgese and Ramagopalan say that they hope to see a more pragmatic assessment approach than is set out in the guidelines. Additionally, clearer direction is needed as companies may be “left in the dark” when planning studies, notes Olsen, pointing to the UK’s National Institute for Health and Care Excellence (NICE) as an example of comprehensive guidance on real-world data.
Nationally fragmented uptake
Integration of JCA reports into national reimbursement processes across the EU has yet to be clearly defined and is likely to vary between countries. Guidance published by the EC in 2024 states that Member States must give “due consideration to the published JCA reports”. This leaves a lot of discretion to individual countries, says Natz. To avoid duplication of efforts, “we would have loved to see a mandatory uptake”, he adds. Nonetheless, he is optimistic that Member States will utilise the report since they have also invested time and effort into the JCA processes.
Meanwhile, Morgese is less sure if all countries will incorporate the JCA reports into their HTA processes. Countries such as France, Germany, Norway, and the Netherlands have established policies for coordinating the JCA with national HTA submissions. But countries like Romania, Greece, and Hungary have not yet issued payer guidance on how reports will be incorporated, based on documents in the public domain.
It remains to be seen what impact the JCA will have on local-level pricing and reimbursement, and this will take some time to understand, says Hopkinson.
Future directions
The EC is due to conduct a formal review of the JCA process in 2028. However, Natz says changes are needed before then. He argues that companies are deciding whether to launch in Europe in the next few years, and “we need to convince those companies now.”
Olsen agrees that more urgency is needed, particularly to allow companies sufficient time to interact with the assessors. Though, he notes that both the EC and the HTACG are open to receiving feedback and there appears to be a willingness for change.
If implemented effectively, the JCA has the potential to provide a unified voice on the clinical effectiveness of new products in the EU, says Morgese. But there is the risk of it becoming a bureaucratic exercise and an added layer of complexity if not done correctly, says Morgese.
