

The US Food and Drug Administration (FDA) has granted approval for Emmaus Life Sciences' Endari (L-glutamine oral powder) to reduce the severe complications of rare, debilitating and lifelong hereditary blood disorder sickle cell disease (SCD) in adult and pediatric patients aged five and older.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
SCD affects around 100,000 patients in the US and up to 25 million patients worldwide, the majority of which are of African descent along with Latinos and other minority groups.
The L-glutamine oral powder is designed to reduce oxidant damage to red blood cells by improving the redox potential of nicotinamide adenine dinucleotide (NAD), a coenzyme that has been identified as the primary regulator of oxidation.
Emmaus Life Sciences chairman and chief executive officer Yutaka Niihara said: "The approval of Endari is a significant milestone for the sickle cell patient community who has not had an advancement in treatment for nearly 20 years, and which now, for the first time ever, has a treatment option for children.
"Endari reinforces our commitment to discovering innovative therapies that help to improve the lives of people with rare diseases. We thank the FDA for its prompt review and look forward to making treatment available to patients as early as this fourth quarter."
The approval is based on data data from a 48-week, randomised, double-blind, placebo-controlled, multicentre Phase III clinical trial evaluating the effects of Endari compared to placebo on 230 adults and children with SCD.
The data was secured from 298 patients treated with L-glutamine and 111 patients treated with placebo in the Phase II and Phase III trials.
The results showed that the drug reduced the frequency of sickle cell crises by 25% and hospitalisations by 33%.
Additional findings showed a decrease in cumulative hospital days by 41% and lower incidence of ACS by more than 60%.
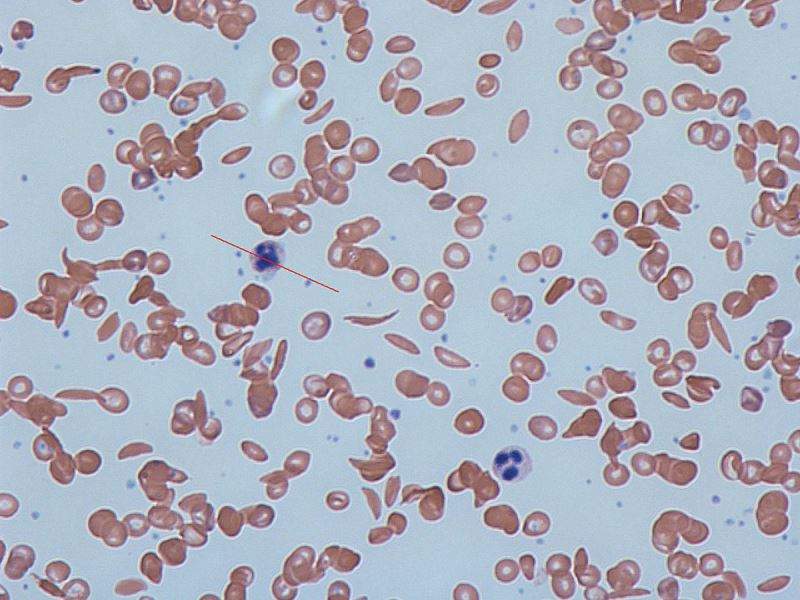
SCD is caused by a genetic mutation in the beta-chain of hemoglobin that distorts red blood cells into crescent shapes.
The disease lowers oxygen levels in the blood and has an extensive impact on morbidity, mortality and quality of life.
Patients with this disease often suffer from debilitating episodes of sickle cell crises, which occur when the rigid, adhesive and inflexible red blood cells block the blood vessels, resulting in excruciating pain.
Image: Sickle cells in human blood: both normal red blood cells and sickle-shaped cells are present. Photo: courtesy of Graham Beards.
