
As biological compounds that are not exact copies of their reference products, biosimilars present far more and newer complexities to regulatory agencies compared to generics.
Although the world’s first biosimilar guidelines were established in 2005 by the European Medicines Agency (EMA), biological compounds have been around for decades. For example, the first approval of a monoclonal antibody was granted by the FDA in 1986 to muronomab-CD3 for the prevention of acute transplant rejection, though this product is no longer available.
Go deeper with GlobalData

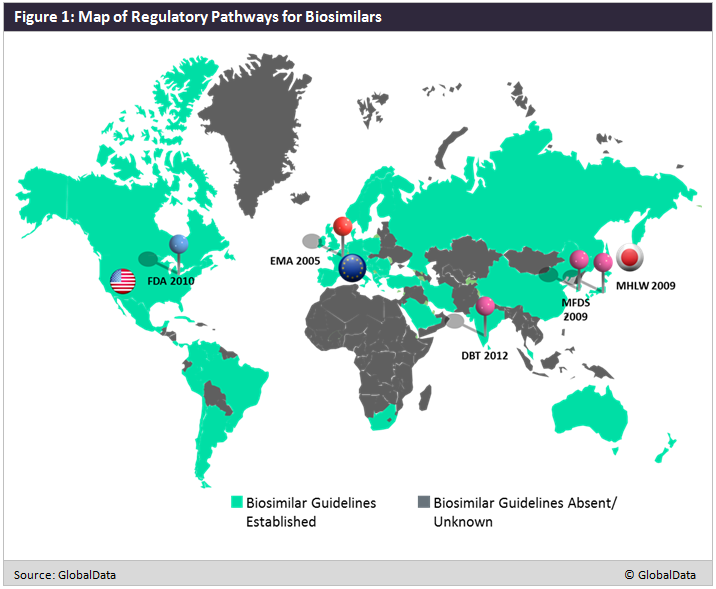
The development of regulatory guidelines sprung from a need for regulatory agencies to define ’biosimilars’ and develop criteria for their clinical development and eventual approval. As depicted in Figure 1 below, most of the developed world has established pathways for biosimilar development, including the EMA in 2005, South Korea’s Ministry Of Food and Drug Safety (MFDS) in 2009, Japan’s Ministry of Health, Labour and Welfare (MHLW) in 2009, the US FDA in 2010, and India’s Department of Biotechnology (DBT) in 2012. Not pictured on the map is the World Health Organization (WHO), which developed its initial guidelines on biosimilarity in 2009.
Over the past decade, regulatory guidance on biosimilars was often critiqued as insufficient and delayed in relation to need, as is evidenced by the fact that three biosimilars were available on the market in India in advance of official guidance. Nonetheless, criteria are continually being re-evaluated, as developers, regulatory bodies, and physicians have a tendency to err on the side of caution as they continue to learn more about these non-identical products, determine whether minute variations will have meaningful clinical impacts, and weigh the clinical impacts against the importance of pricing and access for patients and payers.

Related Reports
GlobalData (2018). Biosimilars in Oncology, to be published
